Contents
1. Overview
Our lab has been involved with research in (i) data compression, a subfield of Information Theory and Signal Processing, which provides the most efficient way to represent information and develops techniques to exploit different kinds of structures in data (ii) bioinformatics, which involves computational methods to organize and analyze biological and clinical data, and (iii) network science, which analyzes phenomena using deterministic and probabilistic graphs. A summary of our selected contributions in these areas is shown below (Fig. 1).
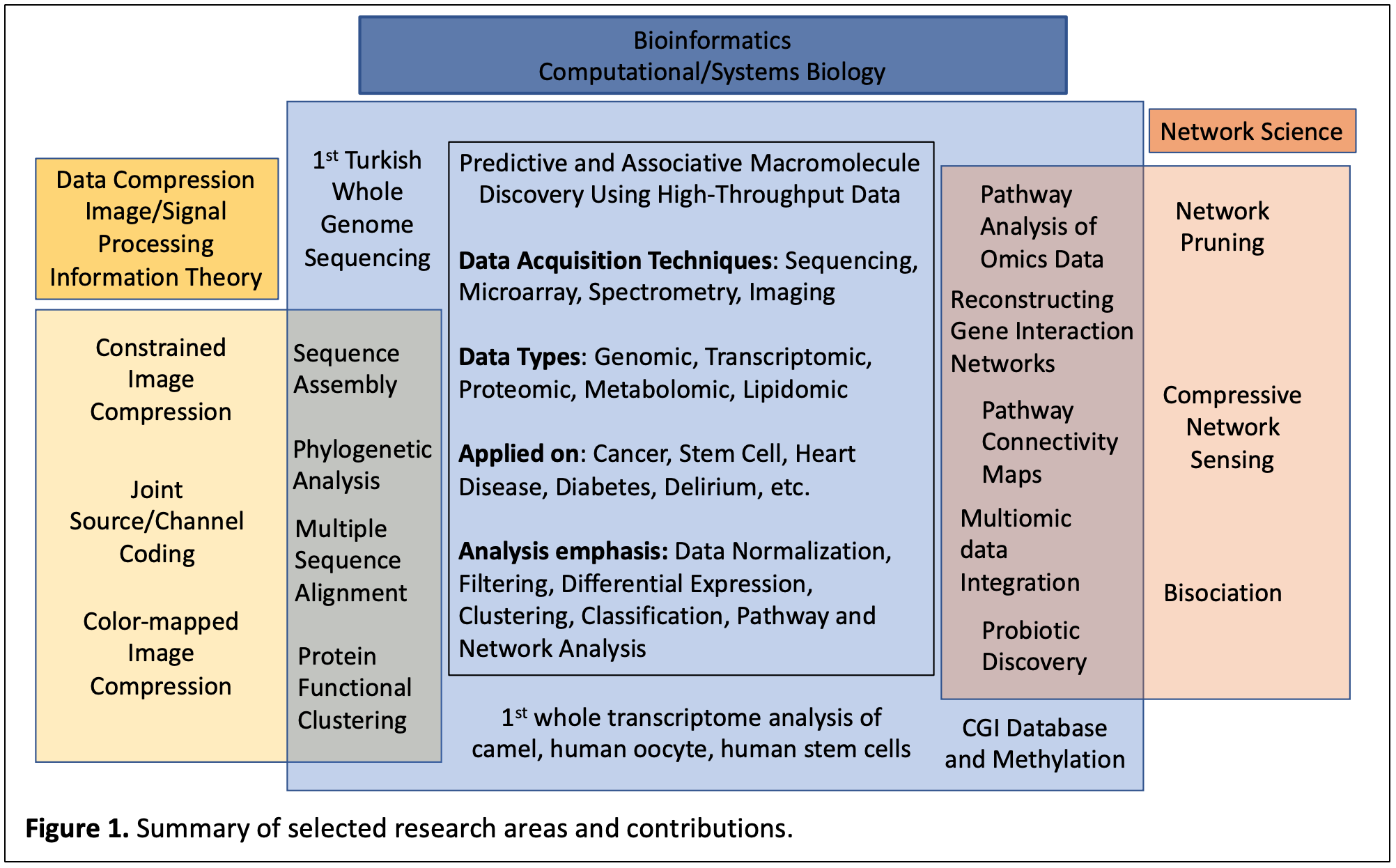
Our earlier work was in the areas of image compression and joint source/channel coding, which combines error correction with data compression. With the availability of genomic sequences, the lab’s interest shifted into bioinformatics by trying to understand how information is organized in DNA sequences. Using Information Theory, we developed characterizations of this organization, with applications to fragment assembly and phylogeny reconstruction.
With the technological advancements in molecular profiling the lab focused on management and analysis of high-throughput biological data (HTBD) from the genomics, transcriptomics, proteomics, metabolomics, and lipidomics domains. These types of data come from different biological sources e.g., tissue, serum, exosome, cerebral spinal fluid (CSF), and acquisition techniques e.g., microarray, DNA/RNA sequencing, imaging, etc. As part of the Bioinformatics Core at Beth Israel Deaconess Medical Center’s (BIDMC) Genomics Center, Harvard Medical School, the core was managed to establish computer infrastructure and research web portal (bidmcgenomics), which functioned as a front end for automated experiment design, ordering, data storage and analysis. As part of this portal, we designed databases for various data types including gene microarrays and protein chips in addition to embedded analysis tools. We have developed analytical methods, which have resulted in standalone computer programs for analysis of HTBD.
The lab has contributed in the areas of data normalization, data filtering, differential expression, clustering, classification/prediction, functional, pathway, and systems level analysis of HTBD. Over the years, we have applied such approaches to phenotypes in health and disease, including but not limited to numerous types of cancer, heart disease, diabetes, and stem cells. These efforts have resulted in papers and patents that identify molecules that are either associated with or predictive of these states (biomarker discovery). In addition to HTBD analysis, of note, the lab assumed a leading role in projects that performed the first whole genome sequencing of a Turkish individual, the first large-scale transcriptome assessment of the Arabian camel, the human oocyte, and the human stem cells; and projects that involved other macromolecular sequence-based efforts including metagenomics, protein functional classification, phylogenetic analysis, CpG island characterization and methylation status assessment.
Our overall aim is understanding biological phenomena within the context of networks, along the lines of systems biology. We applied mathematical, statistical, and engineering approaches to identify active known biological pathways given HTBD using probabilistic graph representations. We supplemented this approach to learning new gene regulatory networks rendered from experimental data using external biological knowledge. This approach was extended to whole organism level interaction network (atlas) learning via a divide-and-conquer approach, and to a new framework that generated interaction networks based on multi-omic data. We have also applied network science to modeling known probiotics to identify new and effective multi-species and next-generation probiotics.
2. Ongoing Research Projects
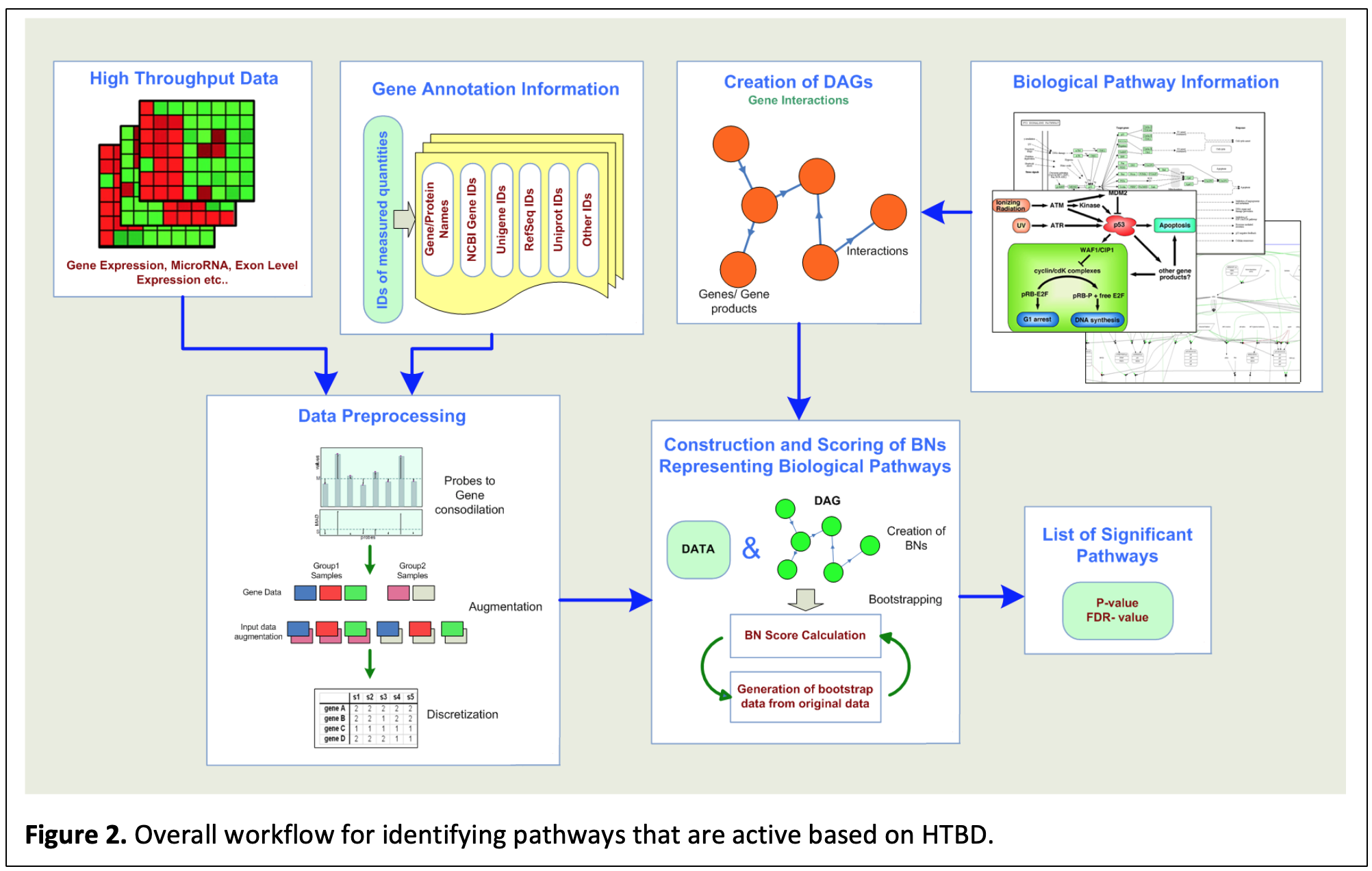
Bayesian Pathway Analysis: Bayesian Networks (BNs), which are directed acyclic graphs (DAGs) representing conditional dependency structure for a set of random variables have been used with increasing popularity over the past 30 years in a diverse range of areas spanning from economics to biology. BNs capture both linear and nonlinear interactions, handle stochastic events in a probabilistic framework accounting for noise, and focus on local interactions, which can be related to causal inference. These properties make BNs viable candidates to be used for analysis of HTBD in a network setting. Most of the existing methods that functionally analyze a set of “interesting” molecules treat them simply as “lists” and perform enrichment analysis based on the over-representation of the functional group (e.g. a pathway) by the input list. We transcended this approach by incorporating the topology, i.e., the connectedness of the molecules in a given pathway to determine its association with the data. Briefly, each pathway is modeled as a BN and the input data is preprocessed to be tested for fitness to the pathways. Following bootstrap and multiple hypotheses testing for statistical significance, pathways that are deemed active based on the given HTBD are identified. Overall approach is summarized in Figure 2 (http://otulab.unl.edu/bpa).
Bayesian Network Prior:
Most of the algorithmic tasks for structure learning to find the graph G that best explains given data D are performed by calculating P(D│G) instead of the true parameter P(G│D).Considering the relation
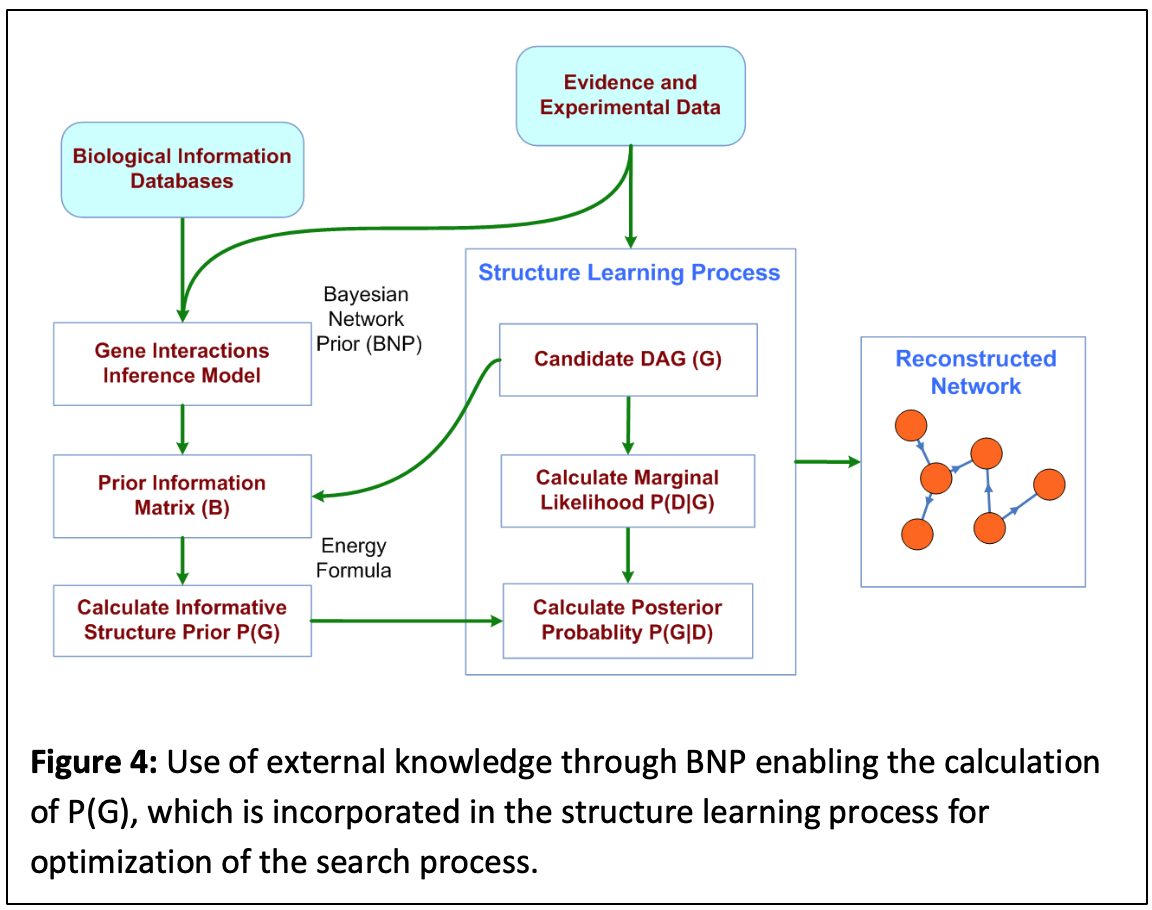
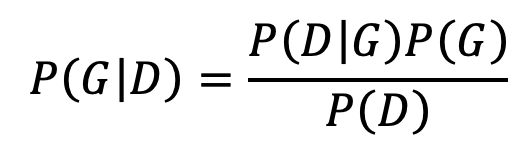 the choice of calculating P(D│G) instead of P(G│D) is justified by assuming a constant value for P(D) and equal probability for all Gs, i.e. a uniform prior structure for all possible candidate DAGs. Although assuming a constant value for P(D) is sound as D is observed, P(G)s are assumed equal for lack of prior knowledge on Gs. We hypothesize that in case of learning interaction networks from HTBD, P(G) can be calculated using external knowledge. Thus, we calculate
the choice of calculating P(D│G) instead of P(G│D) is justified by assuming a constant value for P(D) and equal probability for all Gs, i.e. a uniform prior structure for all possible candidate DAGs. Although assuming a constant value for P(D) is sound as D is observed, P(G)s are assumed equal for lack of prior knowledge on Gs. We hypothesize that in case of learning interaction networks from HTBD, P(G) can be calculated using external knowledge. Thus, we calculate

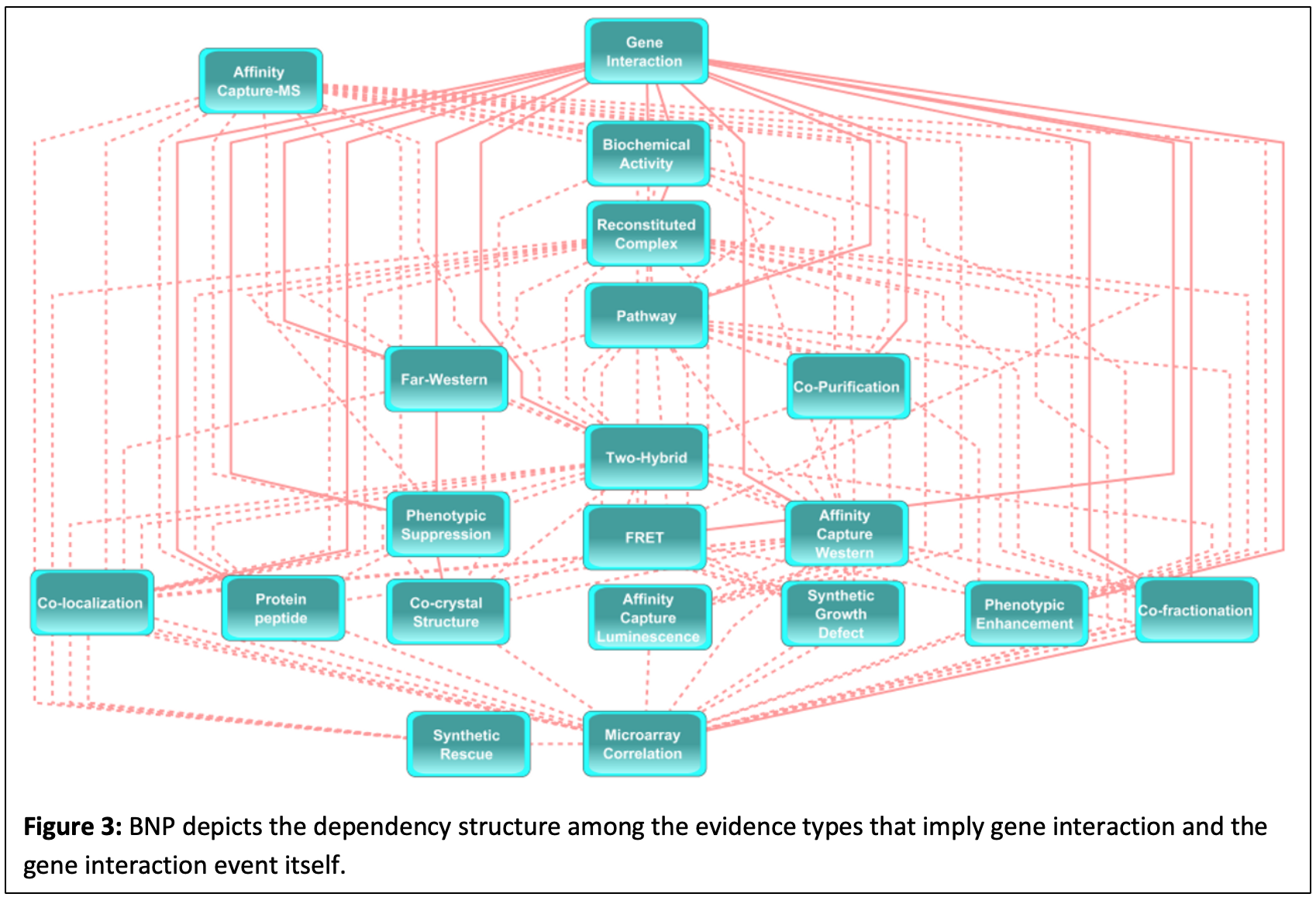
We determine the interaction probability of two genes by establishing a BN using existing external knowledge, which is obtained from interaction databases such as BioGrid, Gene Ontology, and KEGG and consist of protein-protein, protein-DNA/RNA, gene interactions and co-expression data from microarrays. We call this BN structure that can be used as a decision-making system if two genes interact with each other, BNP (BN Prior, Fig. 3). BNP consists of one “interaction” node and other nodes representing external knowledge types. The topology of this BN is determined by employing BN structure learning algorithms on existing external knowledge and known gene interactions. BNP is obtained off-line, prior to the application of given HTBD.
In due process, we instantiate BNP with the given HTBD with all possible pairs of genes at a time. Using the outcomes represented by the “interaction” node, we end up with a matrix, B, where bij represents the probability that gene i and j interact given prior knowledge. We use a novel energy function that calculates P(G) for a candidate graph G in the structure learning process using B (both considered as adjacency matrices). Our results show successful application of this approach on synthetic and real data. Fig. 4 represents the overall workflow depicting the use of BNP in interaction network learning (http://otulab.unl.edu/bnp).
Atlas Learning:
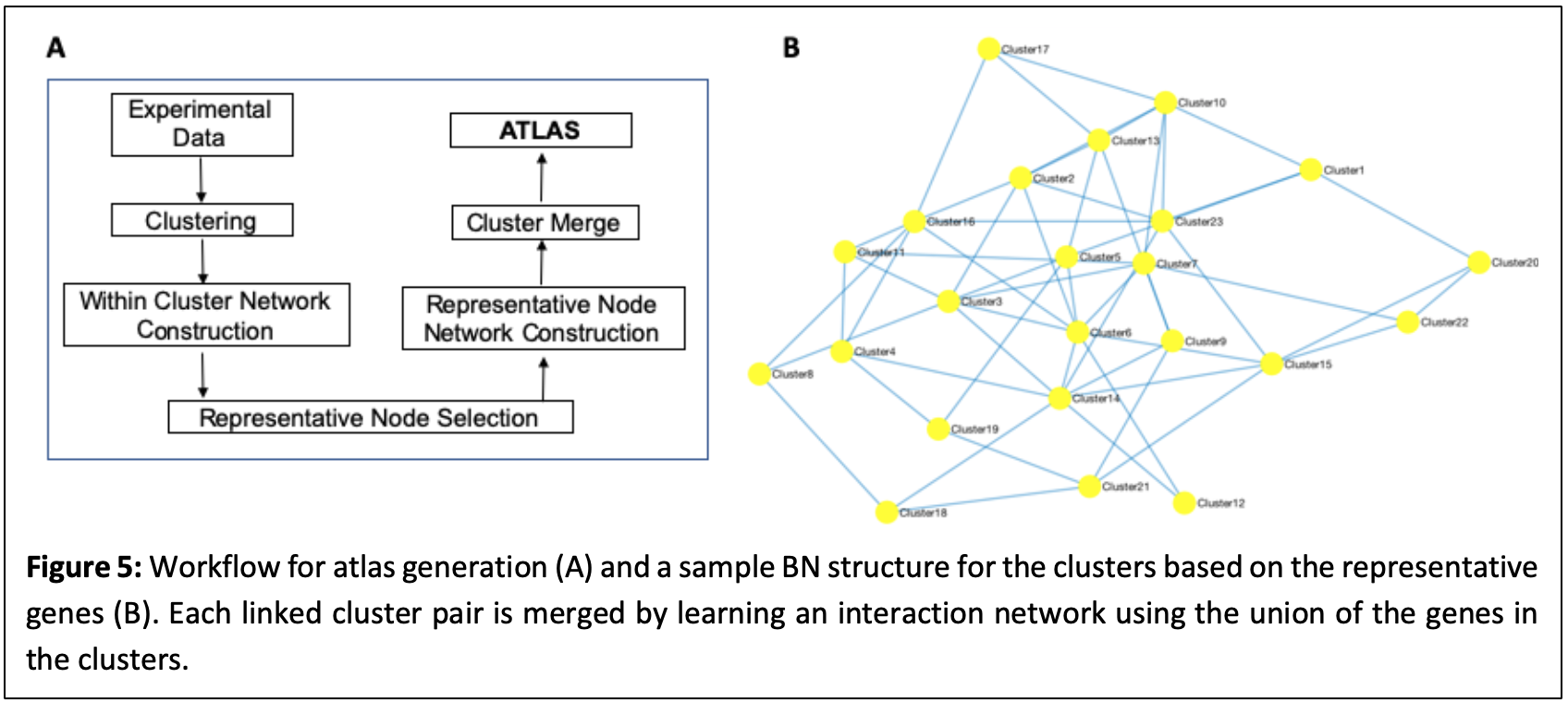
Building gene interaction networks using HTBD is computationally complex and is therefore usually performed on at most a few hundreds of genes. However, biological systems are far more complex and there is need to evaluate the “big picture.” We extended the BNP workflow (Fig. 4) to the complete gene interaction network for an organism (or the interactome atlas), using an iterative network building procedure. First, gene modules are evaluated based on a degree of similarity (e.g. co-expression) and corresponding clusters are generated. Each cluster is represented by one data point (e.g. one gene in the cluster) and a BN is calculated for the representative genes using the BNP framework, which also implies a network for the modules (clusters). Using this graph as a map, connected clusters are used to build interaction networks for the genes involved in the clusters that gives rise to the overall atlas. This approach is summarized in Fig. 5. We have built interactome atlases for gastrointestinal cancer types based on the RNA-seq data obtained from the cancer genome atlas (TCGA). Resulting networks are evaluated based on network parameters (e.g. degree distribution, clustering coefficient), emerging network motifs, and random matrix theory. We have built two supplementary software packages FQStat (http://otulab.unl.edu/FQStat) and KEGG2Net (http://otulab.unl.edu/kegg2net) that accompany the atlas generation project.
Multiomic Data Integration: We have built a database of interactions among multiomic end-points (e.g. proteins, metabolites, and lipids) based on existing external knowledge. Multiomic HTBD is obtained for the same samples and an interaction network is obtained among different omic endpoints using probabilistic graph methods that are further evaluated based on the external knowledge. We specifically applied this approach to multiomics of delirium and evaluated resulting networks for control and delirium samples. We plan to find perturbations in the original multiomic expression values that result in an alignment of the two networks: healthy and delirious, which would imply potential therapeutic interventions.
CpG Island Analysis: We have built a database dbCGI (http://otulab.unl.edu/dbCGI) that, for the first time, identifies, analyzes, and annotates CpG islands (CGIs). CGIs are stretches of DNA in vertebrate organisms with an overrepresentation of the CpG dinucleotide and are naturally unmethylated. DNA methylation is a widely studied epigenetic modification that occurs in the cytosine nucleotide, which results in a passively condensed state of chromatin and is involved in many cellular events, such as gene expression, chromosomal stability, cell differentiation, and carcinogenesis. There have been genes that are identified as methylation-prone and methylation-resistant based on experimental approaches. We developed predictive models that capture this class distinction based on sequence features. We further position the sequence features in the predictive model as determinants that turn a methylation-resistant gene into a methylation-prone gene, similar to a single nucleotide polymorphism (SNP) having a dramatic effect on gene function. The goal is to identify a new set of sequence features that may help explain or predict disease states due to CGIs of certain genes (e.g. tumor suppressor genes) accumulating such features that may lead to methylation.
3. Collaborations
Our lab enjoys a wide array of collaborations with a multitude of internationally recognized scientists.
Towia Libermann (BIDMC; Harvard Medical School): Various projects mostly based on proteomic profiling of biospecimens using SOMAscan, most prominently with applications on pancreatic cancer, NASH, IBD, and Delirium.
Jose Cibelli (Michigan State University): Understanding cellular reprogramming by studying the oocyte, in vitro fertilization, somatic cell nuclear transfer, stem cells, and induced pluripotent stem cells in humans and model organisms such as mouse and zebrafish.
Khalid Sayood (University of Nebraska-Lincoln): Information theory-based approaches in Bioinformatics.
Handan Can (University of Nebraska-Lincoln): Computational methods to identify multispecies and next generation probiotics.
Ed Marcantonio (BIDMC; Harvard Medical School) and Sharon Inouye (BIDMC; Institute for Aging Research, Hebrew SeniorLife; Harvard Medical School): Mulitomic approaches for delirium, dementia, and Alzheimer’s disease.
Ali Nawshad (University of Nebraska Medical Center): Transcriptomic analysis of cleft palate and bone regeneration.
Harland Winter (Masssachussetts General Hospital, Harvard Medical School): Multiomic and metagenomic approaches to identify diagnostic and therapeutic agents for the inflammatory bowel disease (IBD).
Andrew Harms (University of Nebraska-Lincoln): Gene interaction network analysis using compressive sensing.
Kirk Druey (National Institutes of Health): Identification of molecular factors altered in systemic capillary leak syndrome (SCLS; Clarkson disease).
Daniel Muruve (University of Calgary): Serum based biomarker discovery in minimal change disease and membranous nephropathy.
Ben Bleier (Massachusetts Eye and Ear Infirmary; Harvard Medical School): Proteomic and transcriptomic analysis of chronic rhinosinusitis with nasal polyps.
Marsha Moses (Boston Children’s Hospital; Harvard Medical School): Proteomics approaches to understand the interactions of breast cancer-derived exosomes with the blood brain barrier.
4. Funding
| 2002-2003 | NIH/NIDDK; Project No: 5U24DK058739-03; “NIDDK Biotechnology Center”; Libermann (PI); $595,856; Role: Investigator |
| 2002-2007 | NIH/NCI; Project No: PO1 CA92664-03; “Spatial and Temporal Regulation of Angiogenesis”; Dvorak (PI); $141,237; Role: Investigator |
| 2003-2005 | NIH/NIAID; Project No: P01 AI041521; “Costimulation and Cytokines in Tolerance”; Turka (PI); $1,459,884; Role: Investigator |
| 2003-2005 | NIH/NCI; Project No: 1R21 CA108303-01; “Proteomics and Biomarkers for Hepatocellular Cancer”; Afdahl (PI); $100,000; Role: Investigator |
| 2004-2009 | NIH/NCI; Project No: P01 HL076540; “Endothelial Cell Phenotypes in Health and Disease”, Aird (PI); $80,000; Role: Investigator |
| 2005-2007 | NIH/NIAID; Project No: R21 CA107352-01; “Novel Approaches to Gene Profiling in Ovarian Cancer”; Libermann (PI); $86,000; Role: Investigator |
| 2006-2010 | Michigan State University-NAY Project; Project No: MSU 95464; “Direct Dedifferentiation of Primary Somatic Cells”, Cibelli (PI); Role: Consultant |
| 2007-2010 | King Abdulaziz City for Science and Technology; Project No: 26-64; “Camel Genome Project Phase I”; Al-Swailem (PI), Otu (Co-PI); $519,281. |
| 2009-2012 | The Dubai Harvard Foundation for Medical Research; “Analysis of high-throughput genomic data using an integrated approach; Otu (PI); $182,000 |
| 2011-2013 | Istanbul Bilgi University Research Fund; “Human Whole Genome Sequencing” Otu (PI); $25,000 |
| 2011-2013 | The Scientific and Technological Research Council of Turkey; Project No: 111E042; “Bayesian Network Analysis of High Throughput Biological Data: A Systems Biology Approach”; Otu (PI); $85,000 |
| 2016-2019 | NIH/NIA; Project No: R01AG051658; “Advancing the Understanding of Postoperative Delirium Mechanisms via Multi-Omics”; Marcantonio/Libermann (MPI’s), Otu (Co-PI); ~$2.3M |
| 2018-2020 | NIH/NLM; Project No: R21LM012759; “Identification and characterization of interaction atlases in human”; Otu (PI); $443,862 |
| 2018-2024 | NIH/NIA; Project No: P01AG031720; “Delirium, Dementia, and the Vulnerable Brain: An Integrative Approach”; Inouye (PI); “The role of inflammation in the pathophysiology of delirium and its associated long term cognitive decline (Project 2)”; Marcantonio/Libermann (MPI’s), Otu (Co-PI); ~$13.6M |
Pending/In Preparation
- Electrochemical Chip to Profile Circulating miRNA at Attomolar Level with Zero Background
(Submitted as a BRG R01, Saraf – PI, Otu – Co-PI) - Identification of Multi-Species and Next-Generation Probiotics Using Interaction and Association Networks
(NIH R21, Can – PI, Otu – Co-PI) - Increasing the Efficiency of Zebrafish Somatic Cell Nuclear Transfer Using Multi-Omic-Based Methods
(NIH R21, Cibelli – PI, Otu – Co-PI) - Pathway Connectivity Maps: Bisociation of Interaction Networks Using Network Pruning and Compressive Sensing
(To be submitted as an NIH R01, Otu – PI)